近日,《Journal of Chemical Theory and Computation》(化学理论与计算杂志)上刊登了清华大学燃烧能源中心许雪飞课题组的研究工作,论文题目是《Modulation of electric field and interface on competitive reaction mechanisms》(竞争反应机制中电场和界面的调制)。文章通过包含长程静电作用的深度势能分子动力学模拟,系统研究了在电场存在下的体相水中以及水界面处溶剂化甘氨酸的互变异构过程,并深入分析了电场和界面对甘氨酸(去)质子化状态、互变异构机制和动力学的具体影响。
研究背景
在生物体系中,电场效应无处不在。这源于酶活性位点的静电预组织、金属离子流动、带电生物分子(如DNA和蛋白质)的存在、水分子运动,以及外部电场的直接诱导。这些电场能显著调控溶剂分子取向和生物分子的排列、性质及行为,例如改变氨基酸质子化状态、影响肽链合成与聚集、决定蛋白质折叠模式,甚至促进化学反应和生物催化。然而,电场对生物分子基本过程(如质子转移)的具体调控机制尚缺乏系统认知。
甘氨酸作为最简单的氨基酸,其两性离子态([Z])与中性态([N])间的互变异构是生物体系中质子迁移的模型反应。前期理论研究表明,该过程在溶液中可通过分子内质子转移(Intra-PT)或水合氢离子(H3O+)或氢氧根离子(OH-)介导的分子间质子转移(Inter-PT)两种路径实现。值得注意的是,早期研究曾提出电场可能通过影响水分子取向调控质子转移,但电场如何微观上影响甘氨酸互变异构的热力学、动力学及路径选择仍未知。
另一方面,空气-水界面因其独特的物理化学特性备受关注。近期研究表明,微液滴界面可自发产生约100 mV/Å的强电场,并显著加速多种化学反应。尽管界面电场的存在仍有争议,其潜在效应对化学反应调控的重要性已引发广泛探讨。界面环境是否会通过电场或溶剂效应改变甘氨酸的互变异构行为,尤其是与体相溶液中的电场效应有何异同,是亟待揭示的科学问题。
因此,本研究结合深度势能分子动力学(DPMD)与增强采样技术,系统探究了外部电场(5-10 mV/Å)及空气-水界面对甘氨酸互变异构的调控作用。通过开发融合长程静电相互作用的深度学习势函数(DPLR),实现了电场环境下反应自由能面和动力学过程的精确模拟(视频1),旨在阐明电场与界面如何通过热力学稳定性和动力学能垒影响反应路径选择,为理解生物体系质子迁移及界面催化提供新视角。
视频1 甘氨酸在电场下的DPLR分子模拟视频
研究方法
本研究采用深度势能分子动力学模拟结合增强采样技术,系统探究电场与界面对甘氨酸互变异构的调控机制。核心方法是构建融合长程静电作用的DPLR模型,通过深度Wannier(DW)模型预测原子周围的Wannier中心分布,精确刻画外场诱导的电子极化效应。模型训练基于M06-2X/QZV3P级别的量子化学计算数据,并针对电场环境补充构象样本,最终能量和力预测误差分别低于1.06 meV/atom和49 meV/Å,可靠复现电场下体系的响应行为(图1)。
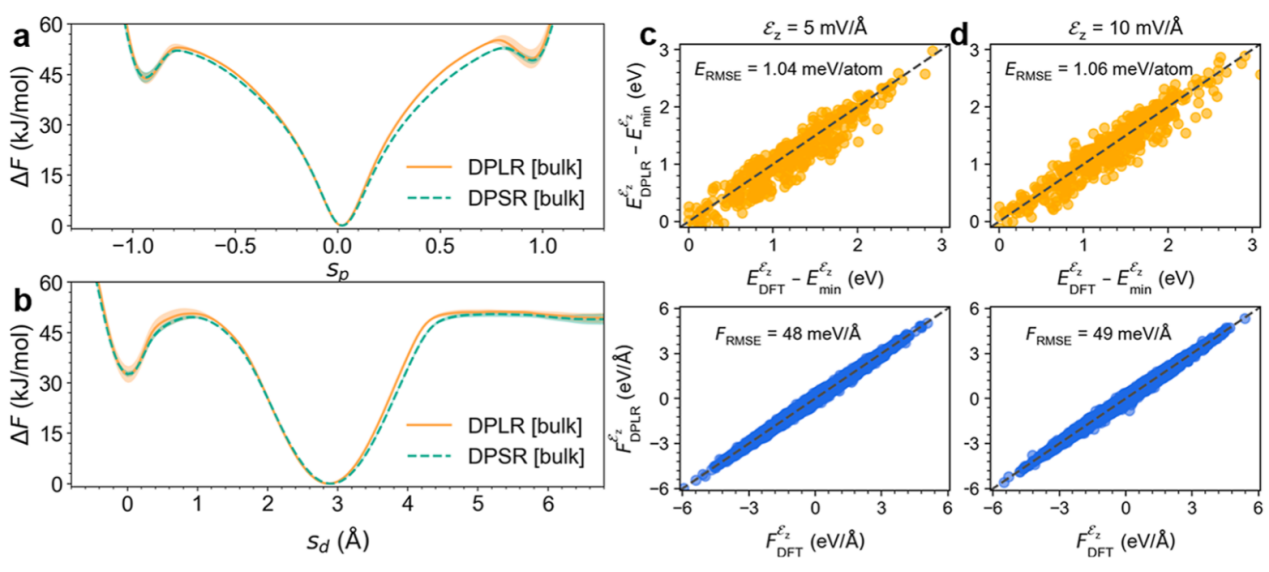
图1. DPLR模型验证 (a,b) 在体相水中,DPLR与DPSR模型计算的甘氨酸自由能曲线比较(以质子化坐标sp或电荷间距sd为函数);(c,d) 电场下(ez =5 和10mV/Å)DPLR模型在测试集上的能量和力误差分布
为解析质子转移反应路径,研究引入即时概率增强采样(OPES)技术,定义质子化坐标(sp)和电荷分离距离(sd)两个关键集体变量。质子化坐标sp量化甘氨酸基团的质子状态变化,电荷分离距离sd则描述离子对的空间分离程度,二者共同构建二维自由能面以捕捉反应中间态与过渡态。通过最小自由能路径分析,明确区分Intra-PT与Inter-PT路径,后者进一步分为H3O+和OH-离子对参与的两种路径。
模拟体系设计涵盖体相溶液与空气-水界面两种环境。体相电场效应通过在立方周期盒子中施加z轴方向电场实现,定量分析电场对自由能面及动力学能垒的影响;界面效应则采用平板水模型,对比界面与体相的热力学稳定性与路径偏好差异。
模型参数、输入文件及数据集通过GitHub(https://github.com/Zhang-pchao/research)与Zenodo(https://doi.org/10.5281/zenodo.14469805)公开。
研究结果
本研究揭示了电场与空气-水界面对甘氨酸互变异构的差异化调控机制(图2)。在电场效应方面,施加5-10 mV/Å的外电场显著重构自由能面:甘氨酸中性态([N])和离子对([C]-OH-/[A]-H3O+)的热力学稳定性提升,其中[N]相对两性离子态([Z])的自由能差降低7 kJ/mol;[N]的稳定由熵减主导(电场迫使极性分子定向排列,降低体系混乱度),而离子对的稳定则受静电效应驱动([C]-OH-自由能降幅达13.4 kJ/mol)。动力学上,电场使[Z]到[N]的三种路径能垒普遍降低,反应速率提升约10倍,并引发关键路径切换:由于电场促进[C]-OH-离子对的长程分离(平均距离增至8 Å),OH-介导的分子间路径(OH--PT)能垒降幅最大(9.8 kJ/mol),取代Intra-PT路径成为主导机制。反向[N]到[Z]反应则因过渡态熵减效应而受抑制。
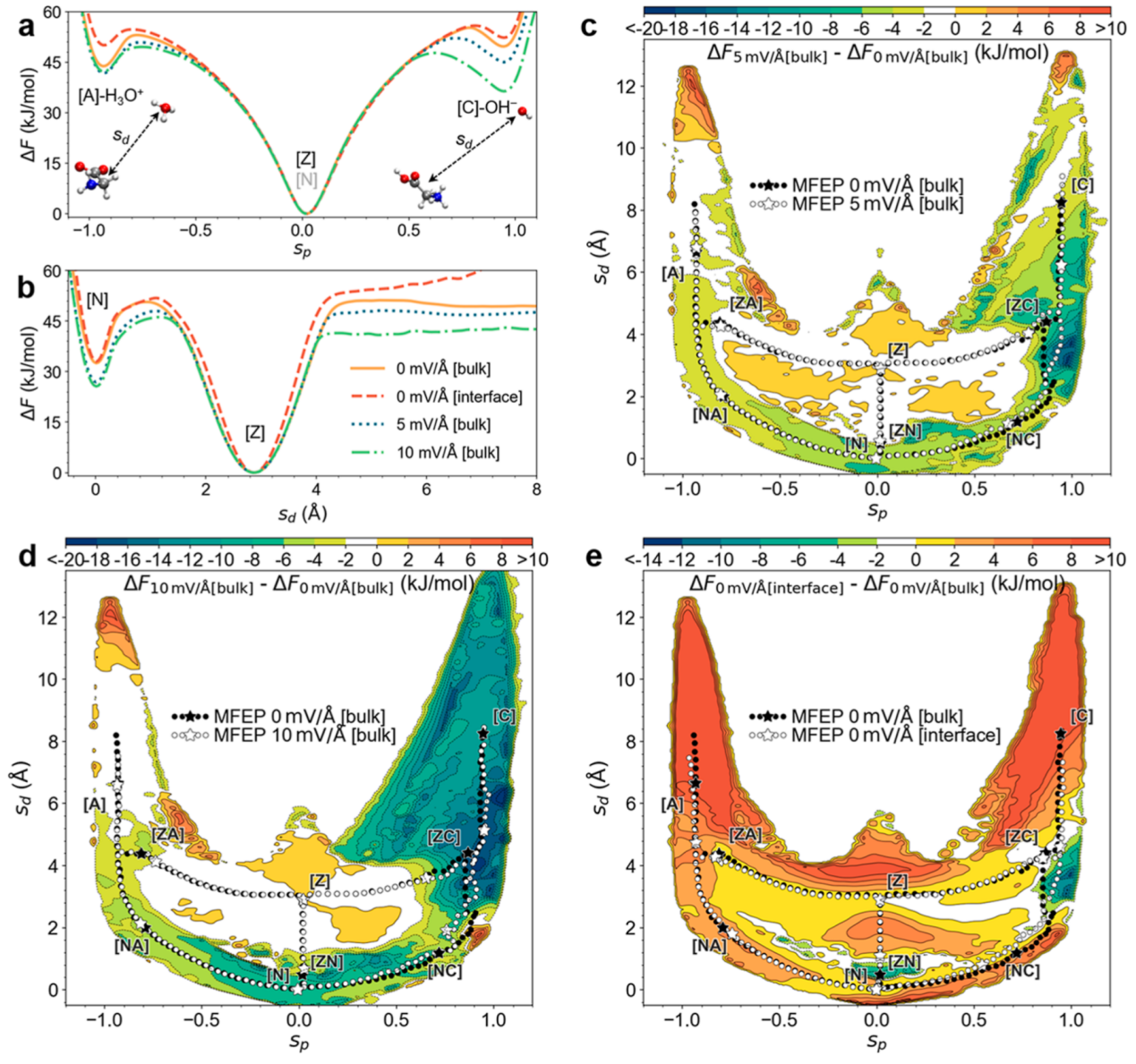
图2. 不同条件下甘氨酸自由能面比较 (a,b) 沿关键反应坐标的自由能一维投影;(c-e)体相电场(5和10 mV/Å)及界面环境下相对体相无电场体系的自由能面差异图(虚线为最小自由能路径,星标为关键结构)
在空气-水界面环境中,界面效应对热力学的调控截然不同。[Z]与[N]之间的平衡自由能差仅变化0.4 kJ/mol,但由于"界面限域效应",离子对稳定性显著降低([A]-H3O+自由能升高5.7 kJ/mol)。这种"界面限域效应"源于甘氨酸与H3O+/OH-离子均倾向于在界面富集,导致离子对分离距离被压缩(分布宽度减小约50%)。动力学上,界面通过部分溶剂化(第一水合壳层水分子减少约30%)降低Intra-PT路径的溶剂化重组能,使其双向能垒降低4 kJ/mol(速率提升5倍)。然而Inter-PT路径因空间约束而大幅减速,形成与电场环境相反的选择性加速模式。
本研究进一步通过机制对比归纳了如下普适规律:电场主要通过熵效应促进分子内质子转移路径,并通过静电效应催化涉及离子对中间体的分子间质子转移路径。界面则因部分溶剂化而加速分子内质子转移路径,并因"界面限域效应"(部分归因于水自离子H3O+/OH-的界面倾向性)而减慢分子间质子转移路径。这些发现为理解电场或界面如何调控多种化学过程的机理和动力学提供了新的视角,也指导了相关应用。
该论文的第一作者为博士生章鹏超,通讯作者为许雪飞副教授。本研究工作得到了国家自然科学基金项目与清华大学山西清洁能源研究院创新基金的资助支持。
文章链接:
https://doi.org/10.1021/acs.jctc.5c00705
ACS Articles on Request link:https://pubs.acs.org/articlesonrequest/AOR-NMVU6VAHH7GKQHZNMPMC
供稿:许雪飞副教授团队
审核:刘有晟、游小清




